
Malattie Rare in Cardiologia: conoscerle per riconoscerle
Nonostante le Malattie Rare che coinvolgono il cuore abbiano ricevuto negli ultimi anni particolare attenzione, soprattutto grazie alla disponibilità di nuovi farmaci, rimangono ancora ampiamente sotto diagnosticate perché poco conosciute e indagate
L’ANMCO ha voluto dare vita quest’anno all’Area Malattie Rare, riconoscendo l’importanza della diagnosi, prevenzione e cura di un gruppo di patologie che merita un approccio più immediato e facilmente percorribile.
Cosa è una malattia rara?
Le malattie rare sono malattie la cui prevalenza non supera, secondo l’Unione Europea, la soglia dello 0,05 per cento della popolazione, ossia 5 casi su 10.000 persone, 1 persona affetta ogni 2000 abitanti. Esistono diverse liste di patologie, tra le quali una delle più utilizzate è riportata da Orphanet, il portale che propone un elenco di più di 6000 patologie rare, sinonimi compresi, destinato a crescere con il progredire della conoscenza e, in particolare, con i progressi della ricerca genetica. Si stima che le malattie rare riguardino nel loro insieme fino al 5% della popolazione mondiale. Secondo la rete Orphanet Italia, nel nostro Paese i malati affetti da patologie rare sono circa 2 milioni. Nel 70-80% dei casi queste malattie hanno un’origine genetica e colpiscono i soggetti già in età pediatrica. Le Malattie Rare hanno ricevuto recentemente una particolare attenzione a livello internazionale con una Risoluzione delle Nazioni Unite, durante la 76esima sessione dell’Assemblea Generale del 16 Dicembre 2021, sostenuta da 54 Paesi. La Risoluzione è uno strumento importante per rafforzare la comunità mondiale delle persone che vivono con una malattia rara, promuovere e incoraggiare le strategie nazionali e la collaborazione internazionale, favorire l’inclusione e la partecipazione nella società delle persone affette e delle loro famiglie e migliorare i risultati sanitari e sociali con le cure e il supporto adeguati.
Quali sono le malattie rare di interesse cardiologico?
Nell’ambito delle Malattie Rare l’apparato cardiovascolare può essere coinvolto sia esclusivamente che insieme ad altre manifestazioni sistemiche, determinando sia la prognosi che l’aspettativa di vita dei pazienti affetti. Alcune malattie, manifestandosi in età pediatrica anche con un complesso quadro sindromico, richiedono un approccio multidisciplinare e il supporto di un cardiologo pediatra. Altre malattie, sia genetiche con esordio tardivo che acquisite, si manifestano in età adulta e vengono all’osservazione del cardiologo come primo specialista a cui va il ruolo di sospettarle clinicamente e approcciarle attraverso un corretto iter diagnostico, al fine di poter intraprendere quando possibile un trattamento specifico. In un tentativo di schematizzazione e semplificazione in base alle competenze specialistiche e di interesse, le Malattie Rare con coinvolgimento del sistema cardiovascolare sono suddivisibili in tre aree: le Cardiomiopatie, le Malattie Aritmiche e le Patologie dell’Aorta.
Cardiomiopatie
Le cardiomiopatie rare possono avere un fenotipo ipertrofico, dilatativo, restrittivo o misto, essere geneticamente determinate o acquisite e colpire soggetti in età adolescenziale o in età adulta. Tra le cardiomiopatie a fenotipo ipertrofico rientrano le forme di cardiomiopatia ipertrofica nell’ambito di un quadro sindromico, come la sindrome di Noonan a trasmissione autosomica dominante, le malattie da accumulo di glicogeno, come la glicogenosi da deficit di LAMP2 o malattia di Danon, la glicogenosi da deficit di maltasi acida e la sindrome PRKAG2, con ereditarietà autosomica dominante. Ne fanno parte inoltre le malattie lisosomiali, tra cui prevale la malattia di Fabry, (Figura 1), una malattia legata al cromosoma X dovuta a deficit di attività dell’enzima alfagalattosidasi A con accumulo di glicosfingolipidi neutri a livello di vari organi e tessuti incluso il cuore.
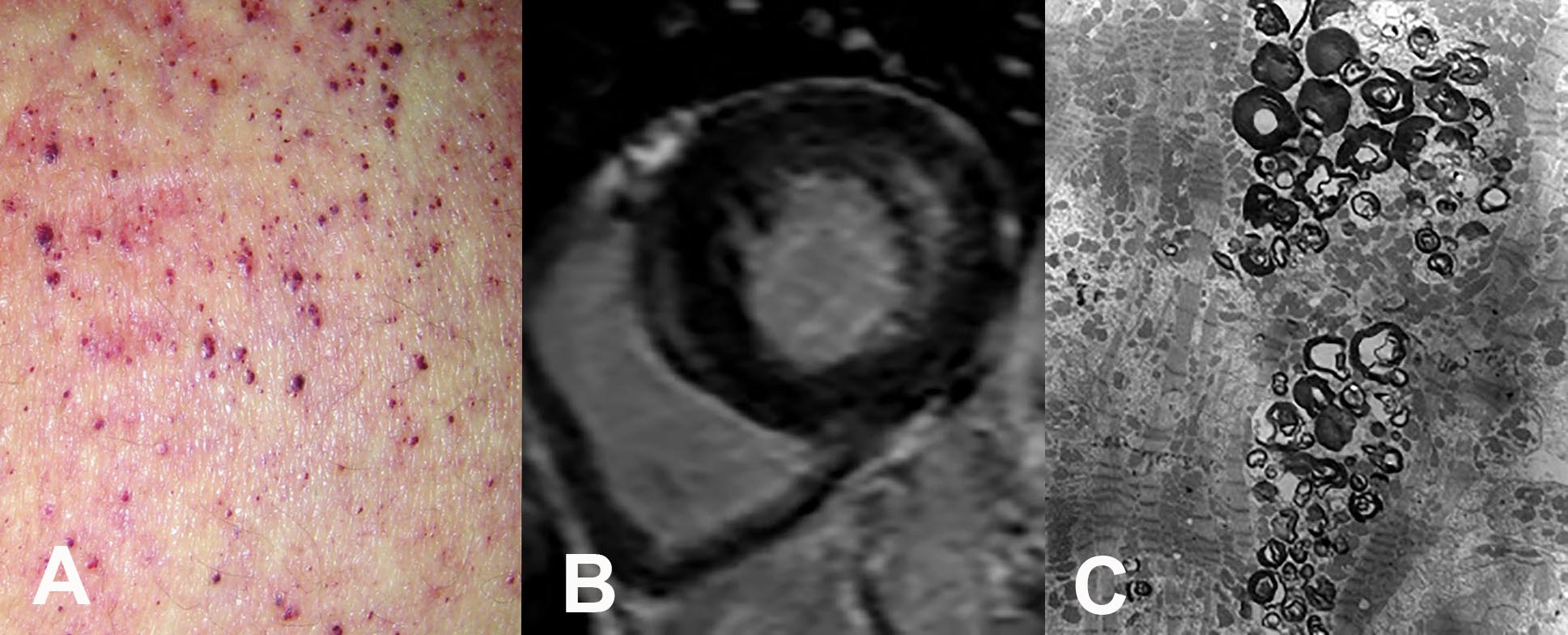
Figura 1 – La malattia di Fabry (o Anderson-Fabry) è una malattia genetica rara trasmessa con il cromosoma X, legata a un deficit dell’enzima alfa-galattosidasi A, che è responsabile del catabolismo dei glicosfingolipidi neutri, con conseguente accumulo del substrato in diversi organi e tessuti, incluso il cuore. È spesso presente una tipica lesione cutanea, l’angiocheratoma corporis diffusum, caratterizzato da lesioni rossastre maculo-papulose (A), e ipertrofia cardiaca (B) dovuta alla deposizione intralisosomiale di glicosfingolipidi (C).
Fanno inoltre parte delle malattie rare a fenotipo ipertrofico/ restrittivo le amiloidosi, sia eredofamiliari (come l’amiloidosi ereditaria da TTR) che acquisite (come l’amiloidosi AA, AL, da transtiretina wild-type), la cardiomiopatia restrittiva familiare e forme restrittive acquisite (es. sindrome di Loeffler, fibroelastosi endomiocardica). Al fenotipo dilatativo appartengono le cardiomiopatie dilatative familiari, come quella da deficit di lamina, alcune mitocondriopatie e sindromi che si manifestano anche con cardiomiopatia dilatativa. Infine, esistono nella classificazione Orphanet delle cardiomiopatie rare quelle definite come “non classificate” tra cui il miocardio non compatto, che è stato associato a mutazioni responsabili sia di cardiomiopatia dilatativa che ipertrofica, la cardiomiopatia di Tako-Tsubo e l’anomalia di Uhl.
Malattie Aritmiche
Fanno parte di questo gruppo tutte quelle malattie rare che si presentano con aritmie cardiache come fenotipo cardiologico esclusivo o prevalente. Un esempio è la sindrome di Brugada, una rara patologia del cuore su base genetica, ad ereditarietà autosomica dominante, legata ad una disfunzione del gene SCN5A, localizzato sul cromosoma 3. Altri esempi sono la sindrome del QT lungo familiare, la sindrome familiare del QT corto, la tachicardia ventricolare polimorfa catecolaminergica, la Displasia/ Cardiomiopatia aritmogena del ventricolo destro, classificata come gruppo di malattie ad ereditarietà autosomica dominante o recessiva ad esordio in età adolescenziale o in età adulta, con una prevalenza stimata tra 1:2.500 e 1:5.000, caratterizzata da alterazioni strutturali (sostituzione fibroadiposa e atrofia miocellulare) del ventricolo destro o di entrambi i ventricoli. Infine, fanno parte di questo gruppo anche malattie genetiche sindromiche con aritmia cardiaca tra le manifestazioni cliniche, come ad es la Sindrome da disabilità intellettiva e aritmia cardiaca da deficit GNB5, una malattia multisistemica a trasmissione autosomica recessiva.
Patologie dell’Aorta
Le patologie aortiche sono a loro volte suddivisibili secondo molti schemi. Una delle caratteristiche più note, tuttavia, è quella di essere inquadrabili in forme sindromiche, riconoscibili quindi attraverso il coinvolgimento caratteristico di più organi, come la sindrome di Marfan, la sindrome di Ehlers- Danlos e la sindrome di Loeys-Dietz (Figura 2). Il fenotipo, tuttavia, può essere multiforme e rendere il riscontro del coinvolgimento aortico potenzialmente difficile e a volte tardivo.
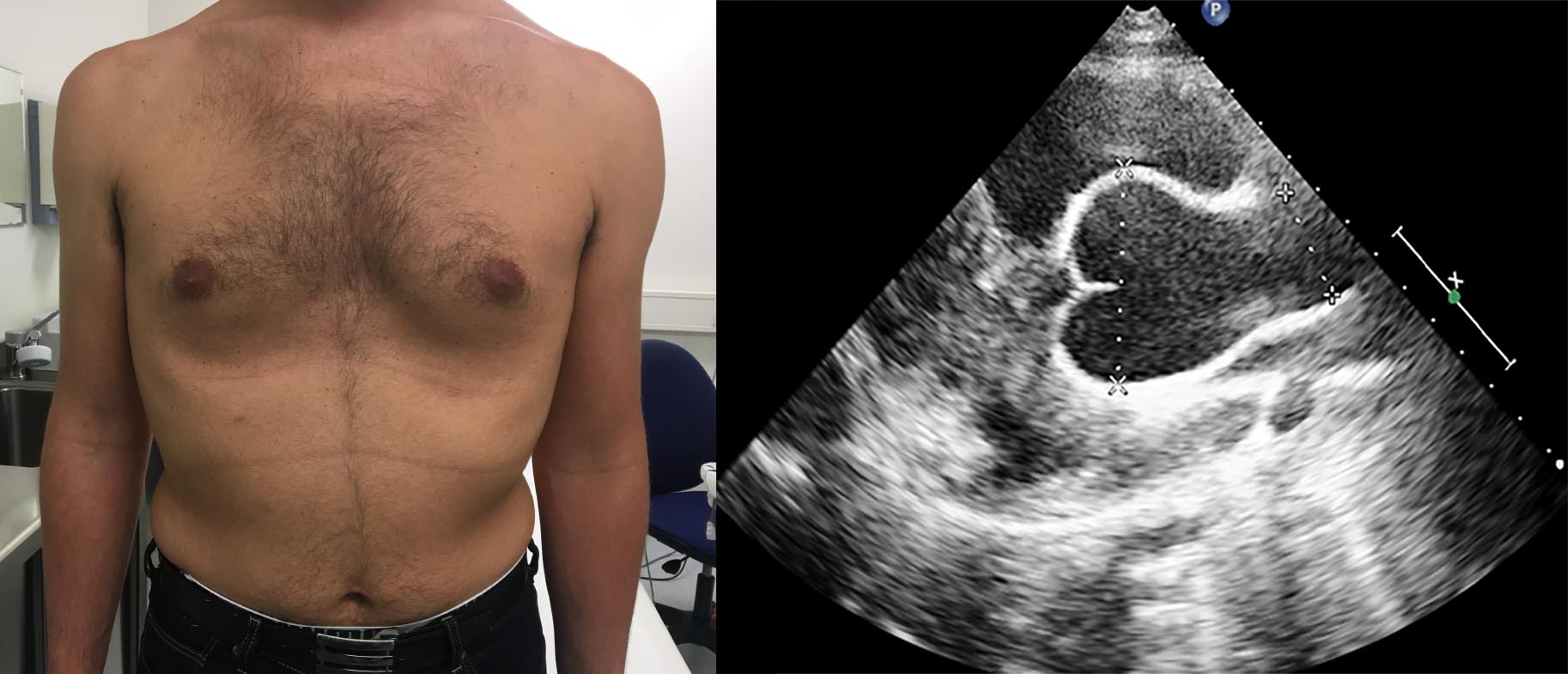
Figura 2 – Le sindromi di Loeys-Dietz rientrano nelle patologie del connettivo e fanno capo ad una serie di mutazioni che danno luogo a 6 diversi sottotipi con possibili caratteristiche fenotipiche quali segni craniofacciali, ugola bifida, craniosinostosi, ipertelorismo, dismorfismi della gabbia toracica, osteoartrite, aneurismi vascolari diffusi e dell’aorta. Riguardano mutazioni a carico dei geni per il fattore di crescita transformante beta (TGFB2 e 3), per i suoi recettori (TGFBR1 e 2) e per le proteine intracellulari che modulano l’attività dei ligandi del fattore di crescita stesso (SMAD 2 e 3).
Come si confronta il cardiologo con le Malattie Rare?
La frequenza con la quale ci confrontiamo con le malattie definite rare è sensibilmente cambiata grazie ad un’informazione più capillare, a test molecolari più facilmente accessibili, e alla revisione delle definizioni ai fini diagnostici che puntano su criteri più facili e di immediata comprensione. I cosidetti “red flags” delle varie malattie guidano il sospetto clinico e orientano i successivi esami diagnostici, inclusi i test genetici. In molte delle sindromi di origine genetica, le manifestazioni sono variabili a causa di una penetranza incompleta della mutazione. Le manifestazioni esterne, campanello di allarme delle manifestazioni interne, possono mancare o essere poco rilevanti. L’assenza di un fenotipo unico rappresentano per il paziente un’incognita e un elemento di ritardo diagnostico potenzialmente fatale. L’approccio alle malattie rare necessita quindi di una maggiore inclusione e condivisione. Il miglioramento della conoscenza da parte del cardiologo delle malattie rare che può incontrare nella pratica clinica è di capillare importanza al fine di porre il sospetto e iniziare una serie di indagini volte al riconoscimento della malattia, con il supporto fondamentale di un team multidisciplinare che in molti casi necessita anche di un pediatra, di un genetista e di uno psicologo, per dare il giusto supporto non solo al probando ma anche ai familiari affetti.

