

Dopo 20 anni dall’introduzione della terapia enzimatica sostitutiva la malattia di Fabry rimane ancora un mistero insoluto
ERT o non ERT? A distanza di 20 anni dall’introduzione della terapia enzimatica sostitutiva ancora non siamo riusciti a identificare prontamente i pazienti affetti dalla malattia di Fabry e soprattutto a curare efficacemente tutti i pazienti
La malattia di Fabry (o Anderson Fabry) è una malattia rara legata al cromosoma X dovuta alla mutazione del gene A GAL, situato sul braccio lungo del cromosoma X, che codifica per l’enzima α-galattosidasi A (α-Gal A). L’assenza o la ridotta attività dell’enzima provoca un accumulo di glicosfingolipidi complessi (globotriaosilceramide, Gb3, e suoi metaboliti, come il LysoGb3), nei lisosomi. Tutte le cellule possono essere coinvolte e il processo provoca un progressivo danno tissutale e insufficienza d’organo. Il coinvolgimento del rene, del cuore e del sistema nervoso centrale ha un impatto critico sulla vita dei pazienti, riducendo l’aspettativa di vita sia dei maschi emizigoti che delle femmine eterozigoti. I fenotipi clinici sono suddivisi in forme classiche e non classiche a esordio tardivo. La malattia classica ha esordio in infanzia-adolescenza ed una grave progressione clinica (cardiomiopatia a fenotipo ipertrofico, insufficienza renale, ictus) nella terza o quarta decade di vita. La forma non classica si caratterizza per un esordio in età adulta ed una progressione del danno più lenta con coinvolgimento clinico esclusivo o prevalente a carico di un organo, ed in particolare il cuore, rientrando nella diagnosi differenziale dei fenotipi ipertrofici. Le caratteristiche cliniche delle femmine sono condizionate dalla trasmissione legata al cromosoma X e dall’inattivazione dell’X wild type o dell’X mutato, con interessamento variabile da organo ad organo e da cellula a cellula nello stesso organo.
La malattia di Fabry prima della terapia enzimatica sostitutiva (ERT): dove eravamo
La malattia di Fabry è stata descritta per la prima volta nel 1898 separatamente e in contemporanea da un dermatologo tedesco (Johannes Fabry) e da un dermatologo inglese (William Anderson), che identificarono la lesione cutanea tipica della malattia, consistente in angectasie cutanee rossastre maculo-papulose più diffuse nella zona periombelicale e scrotale (Figura 1).
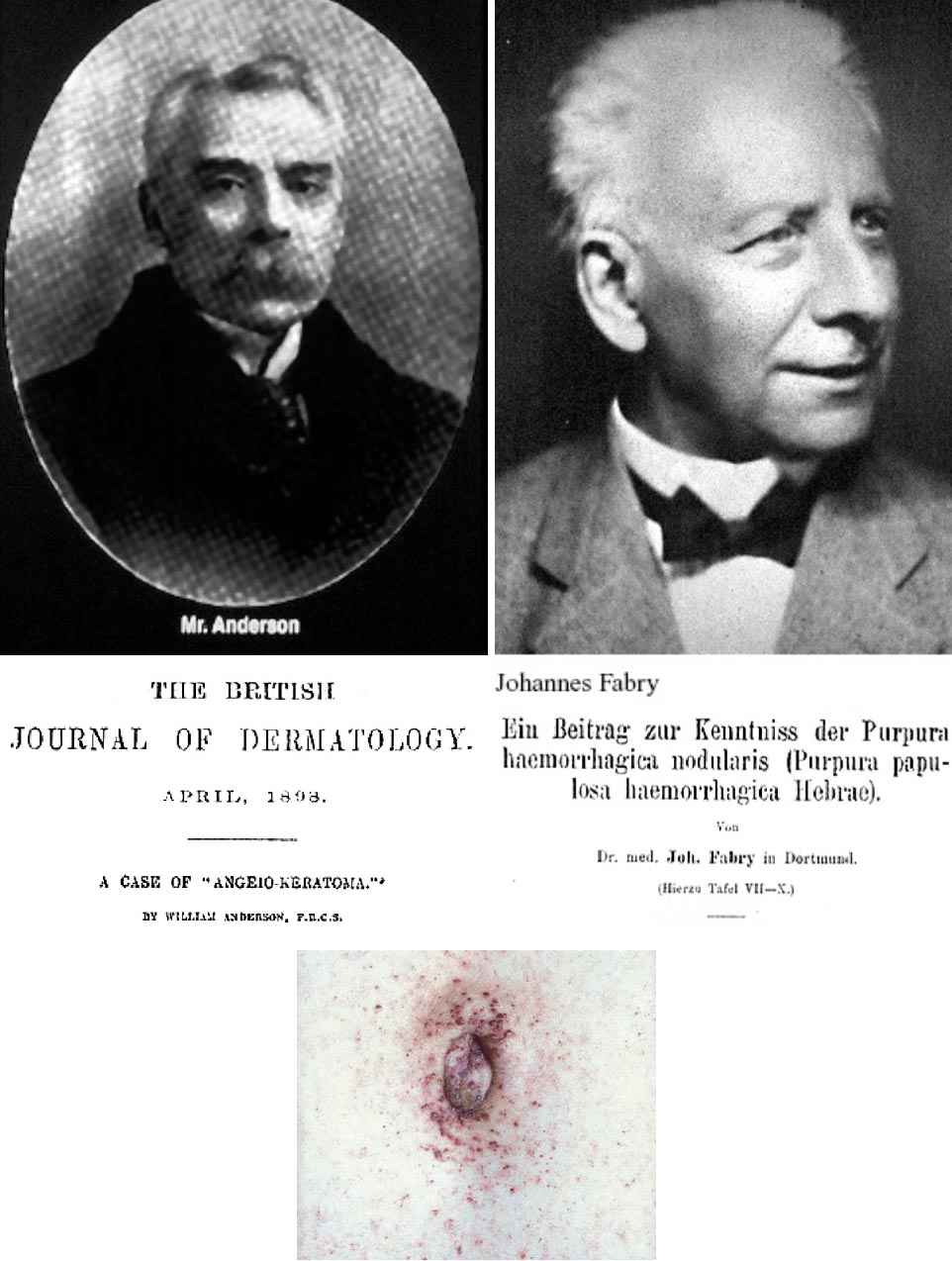
Fabry chiamò questa lesione ‘angiokeratoma corporis diffusum’ ma Anderson riferì della presenza di proteinuria ipotizzando una condizione sistemica generalizzata, probabilmente causata da una malattia vascolare diffusa. Per i successivi 50 anni diversi casi furono riconosciuti e riportati su riviste di dermatologia. Intorno alla metà degli anni ‘50 apparve chiaro come la malattia era in realtà un disturbo da accumulo generalizzato. Intorno alla metà degli anni ’60 furono isolati da organi di pazienti con malattia di Fabry due glicolipidi (ceramide dihexoside e ceramide triesoside) confermando il suggerimento che si trattasse di una malattia da accumulo intralisosomiale di lipidi. Si evidenziò alla microscopia elettronica come questi depositi formassero lamelle disposte concentricamente o in pile parallele; le lamelle erano spesso racchiuse da membrane limitanti mostrando una spaziatura periodica da 40 a 45 Å. Successivamente fu dimostrato che i tessuti di pazienti con malattia di Fabry erano carenti di un enzima idrolitico che scinde il galattosio terminale dalla triesosilceramide. Negli anni successivi si identificò l’enzima alpha-galattosidasi A, si definì il tipo di ereditarietà legata al cromosoma X e furono descritti casi classici di malattia in maschi emizigoti con manifestazioni sistemiche (consistenti in acroparestesie, angiocheratoma, ipoidrosi, opacità corneali e malattia vascolare progressiva del rene, del cuore e del sistema nervoso centrale), di varianti atipiche a insorgenza tardiva e di donne affette dalla malattia nonostante emizigoti. Lo sviluppo di metodiche di sequenziamento genico all’inizio degli anni ‘90 ha permesso poi di isolare l’intera sequenza genomica codificante per A-GAL, ha fornito informazioni fondamentali sulla struttura del gene e della proteina enzimatica.
La malattia di Fabry durante ERT: dove siamo
Fino a prima dell’introduzione della terapia enzimatica sostitutiva l’unico possibile trattamento della malattia di Fabry era la sostituzione d’organo più gravemente colpito, e in particolare il rene. Tuttavia, nei casi descritti in letteratura di trapianto renale appariva evidente come la produzione di enzima ottenuta non era sufficiente a correggere il difetto enzimatico della malattia. Negli anni sono stati fatti tentativi di sopperire all’enzima mancante con infusione di plasma normale per fornire l’enzima attivo o di enzima purificato da tessuto placentare umano, con promettenti risultati in termini di riduzione del substrato circolante. Ma è il 2001 che rappresenta una pietra miliare nella storia della malattia di Fabry, con la commercializzazione della terapia enzimatica sostitutiva con due enzimi di sintesi, l’agalsidasi alfa prodotta dall’attivazione genica nei fibroblasti umani e somministrata alla dose di 0,2 mg/kg per via endovenosa a settimane alterne (Replagal, Takeda/ Shire) e l’agalsidasi beta, un prodotto ricombinante di cellule ovariche di criceto cinese alla dose di 1 mg /kg a settimane alterne (Sanofi/Genzyme). Dopo circa 20 anni di esperienza, sembra che l’ERT abbia cambiato l’approccio ai pazienti con malattia di Fabry e modificato almeno in parte la storia naturale della malattia. È ormai evidente che se la ERT viene iniziata precocemente riesce ad arrestare o a rallentare notevolmente i danni d’organo dovuti alla deposizione di globotriaosilceramide. Al contrario se la ERT viene iniziata quando si è già instaurata ipertrofia ventricolare sinistra o insufficienza renale, non si assiste mai a una regressione della malattia e il risultato che si può ottenere è una stabilizzazione del danno d’organo. C’è ancora qualcosa che ci sfugge nella comprensione dei meccanismi di danno di questa malattia. La risposta all’ERT è raramente completa molto probabilmente per una combinazione di ragioni:
- L’enzima ha una capacità di penetrare i tessuti e di raggiungere le cellule limitata, ad esempio non supera la barriera ematoencefalica, e anche nei tessuti dove penetra probabilmente non arriva in quantità adeguata a livello cellulare.
- Alcuni pazienti sviluppano anticorpi antifarmaco con effetto neutralizzante riducendo l’efficacia della terapia.
- Con l’avanzare della malattia l’aumento della fibrosi cardiaca e renale limitano gli effetti benefici della riduzione dell’accumulo.
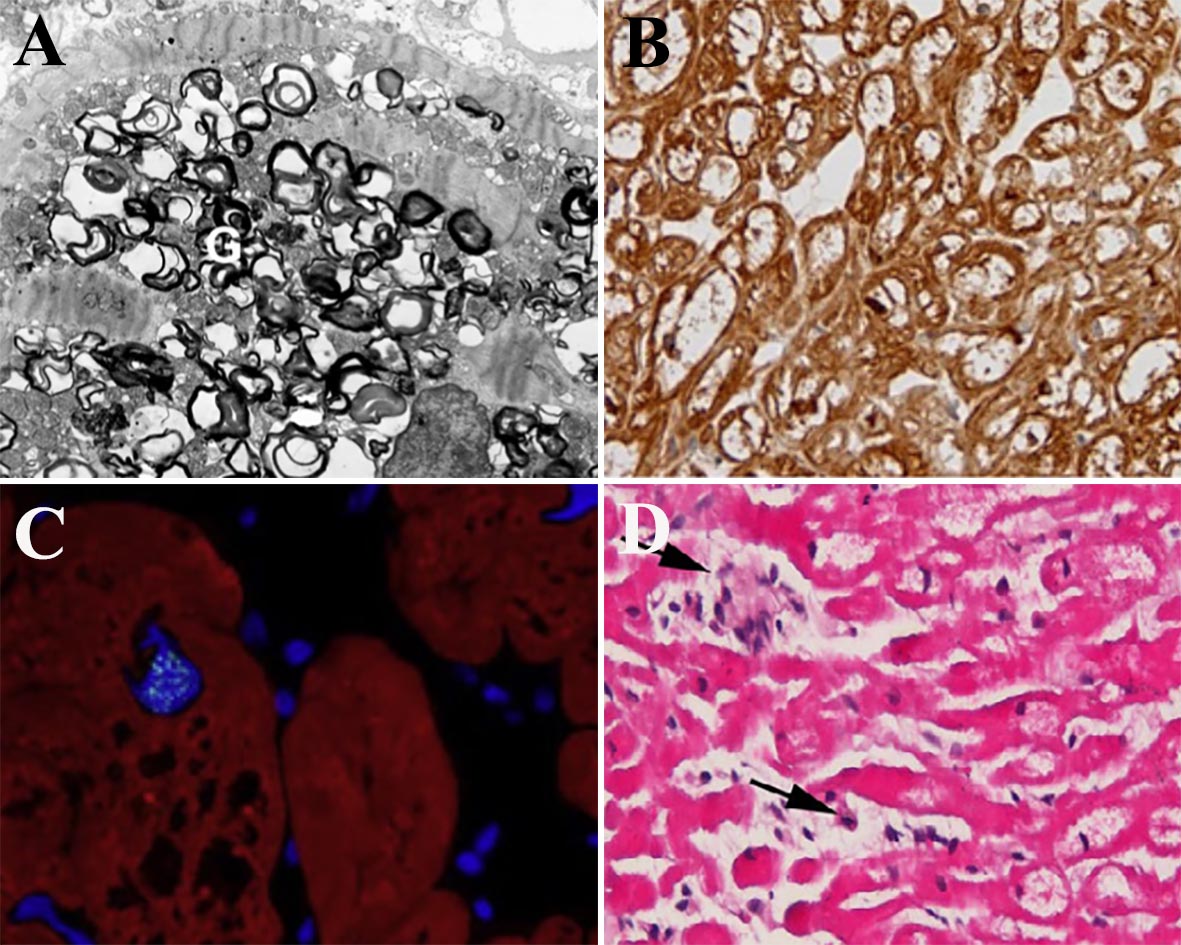
- La deposizione di GB3 a livello degli organi affetti non è solo un fenomeno passivo che aumenta la rigidità delle cellule ingolfate da materiale di accumulo ma ha anche un ruolo attivo. A livello cardiaco l’accumulo intracellulare di GB3 determina infatti l’attivazione di alcuni pathway intracellulari che portano ad apoptosi, stress ossidativo, infiammazione, ipertrofia, che complicano la malattia e limitano la risposta alla ERT (Figura 2).
La malattia di Fabry e le nuove prospettive terapeutiche: dove stiamo andando
La mancanza del successo sperato della terapia enzimatica sostitutiva è stato il trampolino di lancio per la ricerca di nuovi approcci terapeutici. Negli ultimi anni sono stati sviluppati o sono in via di sviluppo nuovi farmaci, che offrendo approcci nuovi e diversi, si prefiggono di agire in maniera più risolutiva sulla malattia. È stata resa recentemente disponibile per il trattamento di prima linea della malattia di Fabry una molecola farmacologica di chaperone nei pazienti Fabry con varianti geniche di A-GAL suscettibili di trattamento. Le mutazioni suscettibili sono associate ad attività enzimatica residua documentata in test in vitro: circa il 30-35% dei pazienti ha un mutazione suscettibile. Migalastat (Galafold, Amicus Therapeutics) è un analogo del galattosio terminale di GB3 in grado di stabilizzare forme suscettibili dell’enzima α-GAL A, aumentandone l’attività e si assume per via orale a giorni alterni. I primi studi disponibili hanno documentato un indice di massa ventricolare sinistra ridotto nei pazienti trattati. Un altro approccio terapeutico ancora in via di sperimentazione in studi di fase 3 è la terapia di riduzione del substrato, che mira a ridurre la produzione di GB3 accumulato. È un approccio completamente diverso al trattamento della malattia, che agisce sulla glucosilceramide sintetasi, l’enzima che catalizza la prima fase della biosintesi dei glicosfingolipidi, bloccandola e garantendo la riduzione della glucosilceramide e della deposizione intracellulare di Gb3. Questi inibitori (es Lucerastat) possono essere assunti per via orale e sono indipendenti dal genotipo. Inoltre, sono in corso studi per la terapia enzimatica con pegunigalsidase alfa, una forma PEGilata dell’agalsidasi di origine vegetale e a somministrazione endovenosa, la cui lunga emivita e ridotta antigenicità sono caratteristiche interessanti. Potrebbe essere somministrato una volta al mese anziché quindicinale. I dati attualmente disponibili hanno dimostrato la capacità di stabilizzare la funzione renale e la massa ventricolare sinistra, ma sono limitati ed è necessario aspettare il completamento degli studi clinici. Infine, la terapia genica potrebbe essere il futuro della malattia di Fabry. È una tecnica che, utilizzando un vettore, virale o non virale, può introdurre una versione correttamente funzionante del gene patologicamente carente. Attualmente sono in atto numerosi studi e nel prossimo futuro potrebbero esserci nuove prove della sicurezza e dell’efficacia di questa terapia. In conclusione la malattia di Fabry è una malattia ancora poco conosciuta e sottodiagnosticata, pur rappresentando una forma potenzialmente trattabile di ipertrofia ventricolare sinistra. Le prospettive per la terapia di questa malattia stanno cambiando e dovremmo essere pronti a implementare possibili innovazioni. Allo stato attuale, l’ERT rappresenta ancora uno strumento eccellente per il trattamento dei pazienti, specialmente in fase precoce. La terapia con chaperoni può essere indicata come prima terapia nei pazienti adulti con mutazioni suscettibili. La terapia di riduzione del substrato, l’enzima PEGilato, la terapia genica e altri preparati enzimatici, sono tutti in fase di sviluppo iniziale e offrono promettenti prospettive per la cura della malattia di Fabry. Noi cardiologi dovremmo quindi continuare sempre più il nostro sforzo di identificare nuovi pazienti affetti.
